Insonnia fatale, la malattia che cancella il sonno a 27 famiglie nel mondo: la cura parte dal Veneto
Il Ca’ Foncello partecipa allo studio dell’istituto Mario Negri di Milano per creare una biobanca di campioni biologici e studiare questa rarissima malattia. Ecco cosa stanno scoprendo i ricercatori sul disturbo che impedisce di dormire
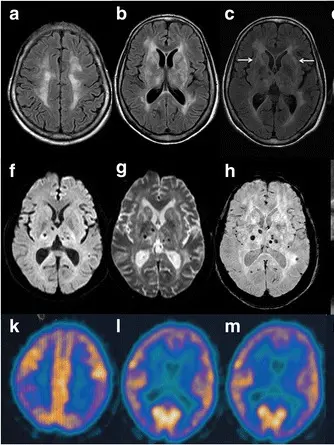
Tanto rara da colpire solo poche persone nel mondo, ma così devastante da cancellare progressivamente una delle funzioni alla base della vita: il sonno. È l’insonnia familiare fatale (Iff), una patologia neurodegenerativa ereditaria causata da una mutazione del gene Prnp, che appartiene al gruppo delle malattie da prioni.
Un disturbo rarissimo che è stato individuato in 27 famiglie di tutto il mondo, due delle quali italiane e una di queste è trevigiana. A livello globale sono noti una sessantina di casi con un’incidenza di una persona ogni milione di abitanti all’anno.

Non esiste ancora una cura, ma proprio da Treviso (dove la malattia è stata individuata per la prima volta negli anni Ottanta) arriva oggi un contributo destinato ad alimentare la ricerca internazionale con l’intento di avvicinare la comunità scientifica alla comprensione dei meccanismi della malattia e poter arrivare a un rimedio.
L’ospedale Ca’ Foncello sarà infatti uno dei protagonisti dello studio promosso dall’Istituto di Ricerche Farmacologiche Mario Negri di Milano, che intende creare una biobanca di campioni biologici provenienti da persone a rischio di sviluppare la malattia per una familiarità conclamata.
La donazione
Il progetto è stato approvato dalla direzione dell’Ulss 2 e sarà sostenuto economicamente dall’Associazione Familiari Insonnia Fatale Familiare (Afiff) di Treviso, che finanzierà per i prossimi tre anni le attività di arruolamento e follow-up dei pazienti, nonché i costi del personale e le spese per reagenti, provette, spedizioni e conservazione dei campioni biologici che verranno prelevati ai candidati. Il tutto grazie a una donazione di 8.500 euro.
Cosa sappiamo
«L’insonnia familiare fatale è una malattia tanto rara quanto inesorabile. La mutazione genetica determina la produzione di una proteina anomala che si accumula nel cervello, colpendo in particolare il talamo, la struttura deputata al controllo del ritmo sonno-veglia» spiega il dottor Francesco Cinetto, immunologo clinico e ricercatore, responsabile locale dello studio e referente dell’Ulss 2 per il Centro Malattie Rare.

Progressivamente il paziente perde la capacità di dormire, mentre compaiono alterazioni cognitive, disturbi motori, disfunzioni del sistema nervoso autonomo e un rapido decadimento neurologico. L’esordio avviene quasi sempre in età adulta e, una volta comparsi i primi sintomi, la sopravvivenza varia da sei mesi a due anni. Oggi la diagnosi genetica consente di identificare i portatori della mutazione anche molti anni prima dell’insorgenza della malattia, aprendo scenari fondamentali per la ricerca di una cura.
L’apporto trevigiano
È proprio su questa finestra temporale che si concentra lo studio del Mario Negri, al quale Treviso offre un contributo determinante. Il progetto segue infatti le orme dell’esperienza maturata negli anni al Ca’ Foncello, grazie al lavoro avviato dal dottor Ignazio Roiter, che prese in carico uno dei primi casi clinici, instaurando una collaborazione con gli enti di ricerca culminata qualche tempo fa in uno studio finanziato da Telethon.
A raccogliere ora quel testimone è il dottor Francesco Cinetto, come lui stesso spiega: «Questo studio, figlio di un percorso iniziato dal dottor Roiter, punterà ora alla raccolta di materiale biologico per costruire la biobanca. Inizia così una nuova fase dello studio, continuativa rispetto al passato e potenzialmente capace di aprire nuovi scenari».
Il monitoraggio
L’attività entrerà nel vivo dopo l’estate. «Ogni sei mesi» prosegue il medico «effettueremo dei prelievi su un gruppo di circa 25 persone tra portatori della mutazione e familiari dei portatori. Tutti i campioni saranno raccolti al Ca’ Foncello, conservati secondo protocolli rigorosi e inviati al Mario Negri». Non si tratta soltanto di prelievi di sangue. La biobanca comprenderà infatti diversi materiali biologici quali plasma, urine, lacrime e biopsie cutanee
. Campioni diversi, raccolti in modo sistematico nel tempo, che permetteranno ai ricercatori di osservare eventuali modificazioni biologiche che precedono la comparsa della malattia. Come evidenzia il dottor Cinetto: «Grazie alla biobanca gli scienziati potranno indagare il comportamento della patologia quando le persone sono ancora asintomatiche. È un’opportunità estremamente importante per preparare il terreno a future strategie terapeutiche». Il Ca’ Foncello si appresta a essere uno dei cardini della ricerca, per fare luce su una delle malattie genetiche più drammatiche e ancora poco conosciute, in un investimento concreto di speranza per il futuro.
L’intervista
Nella lotta alle malattie da prioni che colpiscono il sistema nervoso, il caso Treviso può essere una chiave di volta per la ricerca internazionale. Questione di numerosità di pazienti e di informazioni a disposizione. Come spiega il dottor Roberto Chiesa, responsabile del laboratorio di neurobiologia dei prioni dell’Istituto di Ricerche Farmacologiche Mario Negri di Milano, la nostra provincia rappresenta un unicum.

«La concentrazione di casi di Insonnia familiare fatale in un’area come quella di Treviso può essere spiegata dal punto di vista genetico. Quello che rende questa situazione scientificamente molto rilevante è la possibilità di avere una casistica concentrata e ben seguita nel tempo. Questo ci permette di studiare la malattia in modo molto più approfondito rispetto a contesti in cui i casi sono isolati e dispersi».
Nella provincia di Treviso si registra una presenza straordinaria di casi di Insonnia familiare fatale (Ffi), una malattia prionica rarissima. Come si spiega?
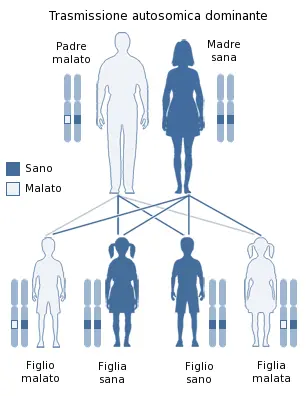
«L’insonnia familiare fatale è una malattia ereditaria legata a una mutazione del gene Prnp. Questa mutazione viene trasmessa in modo autosomico dominante (significa che il gene difettoso prevale sul gene sano dell’altro genitore, con un’elevata probabilità di ammalarsi per i figli). In questi casi, eventi storici di “effetto fondatore” possono portare alla presenza della mutazione in un numero relativamente alto di individui all’interno della stessa area geografica o delle stesse famiglie. Poter avere accesso ai soggetti interessati, nonché ai portatori asintomatici, è estremamente prezioso, perché ci consente di osservare le fasi precliniche della malattia, capendo come e quando inizia il processo patologico. Questo è fondamentale per sviluppare future strategie preventive o interventi molto precoci».
Questo legame tra il territorio della Marca e la ricerca del Mario Negri si sta concretizzando in un progetto ambizioso: la creazione di una biobanca. In che modo la costituzione di una simile raccolta di campioni biologici potrà aiutare il lavoro in laboratorio?
«Una biobanca è uno strumento davvero centrale per far avanzare la ricerca sulle malattie da prioni. Non si tratta semplicemente di conservare dei campioni, ma di costruire una risorsa organizzata, ben caratterizzata e soprattutto condivisibile nel tempo. Avere a disposizione campioni biologici raccolti in modo sistematico (plasma, siero, urina, saliva, lacrime, tamponi nasali, Dna e altri materiali) ci permette di fare una cosa fondamentale: collegare quello che osserviamo dal punto di vista clinico con ciò che accade a livello molecolare. Questo è particolarmente importante nelle fasi molto iniziali della malattia, quando i sintomi non sono ancora evidenti o sono ancora sfumati».
Data l’ereditarietà dell’Insonnia familiare fatale, che valore ha per il gruppo di ricerca poter procedere con un approccio genealogico?
«Una biobanca longitudinale ci consente di seguire l’evoluzione della malattia nel tempo e quindi di capire meglio la sua dinamica, anche prima della comparsa dei sintomi. E questo è un punto chiave se vogliamo pensare non solo alla diagnosi, ma anche a future strategie terapeutiche. Infine, c’è un aspetto che considero essenziale: la possibilità di condividere questi materiali con la comunità scientifica internazionale. Nelle malattie rare questo fa davvero la differenza, perché solo attraverso la collaborazione possiamo raggiungere numeri sufficienti e applicare approcci tecnologici avanzati».
Finora che cosa conosciamo delle malattie da prioni?
«Sappiamo che le malattie da prioni sono patologie neurodegenerative causate da una proteina, la proteina prionica, che assume una conformazione patologica e innesca un processo di propagazione nel cervello. Un punto molto importante che la ricerca ha chiarito è che la presenza della proteina prionica normale è necessaria per lo sviluppo della malattia. Questo ha cambiato completamente la prospettiva terapeutica, perché oggi possiamo immaginare strategie che mirano proprio a ridurre o modulare questa proteina. Sul fronte diagnostico, abbiamo fatto passi avanti enormi con tecniche come la RT-QuIC– Real Time Quaking Induced Conversion, che ci permettono di identificare la presenza di prioni con una sensibilità e una specificità che fino a pochi anni fa non erano immaginabili».
Quali passi avanti ha fatto la ricerca e quali sono gli scenari terapeutici più prossimi?
«Per quanto riguarda le terapie, ci sono linee molto promettenti, alcune già in fase di sperimentazione clinica. In particolare, strategie mirate a ridurre l’espressione del gene della proteina prionica (PRNP), favorire l’eliminazione della proteina o stabilizzarne la sua forma fisiologica. Non abbiamo ancora una terapia efficace nell’uomo, ma oggi siamo molto più vicini rispetto al passato».
La storia: «Incubo durato fino alla diagnosi»
«Non sapevamo cosa stesse accadendo, ma era chiaro che stesse succedendo qualcosa dentro la nostra famiglia». Erano gli anni Sessanta e la scienza non aveva ancora un nome per quel dramma silenzioso che consumava la vita. Nel piccolo paese della Marca Trevigiana, circondato dai campi di mais, le chiacchiere fiorivano in fretta, bollando quegli strani comportamenti come pazzia: «Quella è una famiglia strana, meglio starci alla larga». Ma la follia non c’entrava.
La storia di Luigi rivive oggi nel ricordo concreto dei parenti, anche se il nome scelto per parlare di lui è frutto della fantasia. «Zio Luigi sembrava aver scambiato il giorno con la notte e non aveva più cognizione di sé. Quando i parenti provavano a parlargli, raccontava di fare le valigie, pur trovandosi immobile sul divano. Oppure gesticolava senza controllo, dicendo di vedersi librare nell'aria».
Non era sonnambulismo, ma una misteriosa e terribile malattia che si sarebbe rivelata fatale: una mancanza di sonno totale, che distruggeva il cervello inondandolo di stimoli come in un’eterna, logorante, veglia. Dopo Luigi, un altro caso e poi ancora un terzo. Il dramma si riaffacciò a metà degli anni Ottanta con lo stesso cognome. Fu la volta di una cugina, Lara (altro nome di fantasia), che aveva appena 36 anni. Del suo caso si occupò il dottor Ignazio Roiter, all’epoca in forza all’ospedale di Treviso, che intuì di trovarsi di fronte a una patologia sconosciuta.
Lara accettò di farsi studiare tra Treviso e Bologna, aprendo la pista scientifica: si trattava di un'encefalopatia da prioni mortale. Sempre gli stessi sintomi, le stesse notti in bianco e un effluvio di discorsi sconclusionati a impedire al corpo di poter riposare.
«I nostri parenti se ne andarono nel giro di un anno dalla comparsa dei sintomi, sfiniti nel corpo e nella mente». Fu nel 1986 che arrivò la diagnosi ufficiale a fare luce su quanto stesse accadendo: Insonnia Familiare Fatale che colpisce 27 famiglie al mondo.
Oggi i discendenti trevigiani affrontano questa eredità con dignità e grande speranza. C’è la doxiciclina per provare a rallentare il decorso della malattia e si guarda con fiducia a due molecole in sperimentazione. Inoltre, la Procreazione Medicalmente Assistita (Pma) permette oggi di avere figli sani, non portatori della mutazione. «Cerchiamo di vivere il più normalmente possibile», spiegano i familiari.
«Gli incontri con le genetiste ci hanno mostrato che con la Pma possiamo avere figli senza la mutazione. Per noi è una boccata d’ossigeno e continuiamo a sostenere la ricerca affinché corra più veloce»
Riproduzione riservata © il Nord Est